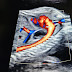
Pulmonary Sequestration vs. Congenital Pulmonary Airway Malformation (CPAM) on Fetal Ultrasonography
Abstract
Pulmonary sequestration (PS) and congenital pulmonary airway malformation (CPAM) are two of the most common congenital lung lesions identified during routine prenatal ultrasonography. While both may present as echogenic thoracic masses, they differ in pathophysiology, vascular supply, and imaging characteristics. Accurate prenatal differentiation using ultrasound, particularly with Doppler, is essential for prognostication and perinatal management. This article reviews and compares the imaging features and clinical implications of PS and CPAM in the fetal period.
1. Introduction
Advances in prenatal imaging have led to the increased detection of congenital lung malformations, with an estimated incidence of 1 in 10,000 to 1 in 35,000 live births1. Pulmonary sequestration and CPAM (formerly congenital cystic adenomatoid malformation) are among the most frequently diagnosed fetal thoracic lesions. Despite overlapping sonographic appearances, distinct features—especially on Doppler imaging—aid in their differentiation.
2. Pulmonary Sequestration (PS)
2.1 Definition and Pathophysiology
Pulmonary sequestration is characterized by non-functional lung tissue that lacks communication with the tracheobronchial tree and receives systemic arterial blood supply, most commonly from the thoracic or abdominal aorta2.
2.2 Types
Intralobar sequestration (ILS): Lies within the normal lobe and shares pleura.
Extralobar sequestration (ELS): Encased in its own pleura and often found below the diaphragm.
2.3 Sonographic Features
Appears as a homogeneous, echogenic mass—typically in the posterior basal lung, often on the left side.
Color Doppler confirms diagnosis by demonstrating a systemic arterial supply from the aorta3.
May cause mediastinal shift or fetal hydrops if large.
CPAM results from abnormal branching of the terminal bronchioles, producing cystic or adenomatous overgrowths of pulmonary tissue that may or may not communicate with the airway. Blood supply is via the pulmonary artery4.
3.2 Classification (Stocker System)
Type I (macrocystic): Dominated by one or more cysts >2 cm.
Type II (microcystic): Smaller cysts <2 cm.
Type III (solid/adenomatoid): Appears solid and echogenic on USG.
3.3 Sonographic Features
May appear cystic, solid, or mixed.
Often presents as a hyperechoic intrathoracic mass, especially in microcystic or solid types.
Color Doppler shows pulmonary arterial supply, not systemic5.
Large lesions may lead to polyhydramnios, mediastinal shift, or hydrops.
4. Key Differentiating Features on Fetal Ultrasonography
Feature
Pulmonary Sequestration (PS)
Congenital Pulmonary Airway Malformation (CPAM)
Echotexture
Homogeneous, echogenic mass
Cystic, mixed, or solid mass
Blood supply
Systemic (aortic) – visible on Doppler
Pulmonary arterial supply
Airway connection
None
May connect with airway
Common location
Posterior/inferior lobe, often left side
Variable, often unilateral
Pleural covering
ELS has separate pleura; ILS does not
Normal pleura
Associated anomalies
Rare; hydrops if large
More commonly associated with hydrops, polyhydramnios
Management approach
Monitor; surgical removal postnatally if symptomatic
May require prenatal or postnatal intervention
5. Diagnostic and Prognostic Tools
Color Doppler ultrasonography is essential to distinguish vascular supply.
CPAM Volume Ratio (CVR) is used to monitor lesion growth and predict risk of hydrops (CVR > 1.6 suggests higher risk)6.
MRI may be used postnatally or in complex cases for better lesion delineation.
6. Management and Outcomes
6.1 Prenatal
Serial ultrasound monitoring.
Maternal corticosteroids (e.g., betamethasone) may be considered for large lesions causing hydrops.
Fetal interventions like thoracoamniotic shunting may be indicated for macrocystic CPAM with mediastinal compression.
6.2 Postnatal
Elective surgical resection is the standard for symptomatic lesions or those at risk of infection or malignancy.
Asymptomatic cases may also undergo resection due to potential complications.
7. Conclusion
Differentiating pulmonary sequestration from CPAM on fetal ultrasound is vital for optimal prenatal counseling and postnatal management. Doppler imaging, lesion location, echotexture, and vascular pattern provide key diagnostic clues. With proper surveillance and intervention, outcomes for most affected fetuses are favorable.
References
Let me know if you'd like a PowerPoint version or a PDF format of this article, or if you want help adding clinical images or diagrams.
Footnotes
Lee EY, Boiselle PM, Cleveland RH. Multidetector CT evaluation of congenital lung anomalies. Radiology. 2008;247(3):632-648. ↩
Adzick NS. Management of fetal lung lesions. Clin Perinatol. 2009;36(2):363–376. ↩
Stocker JT. Congenital pulmonary airway malformation: A new name and an expanded classification of congenital cystic lung lesions. Histopathology. 2002;41 Suppl 2:424–431. ↩
Hubbard AM, Crombleholme TM, Adzick NS. Prenatal MRI evaluation of congenital lung lesions. Semin Pediatr Surg. 2003;12(1):29–33. ↩
Crombleholme TM, Coleman B, Hedrick H, et al. Cystic adenomatoid malformation volume ratio predicts outcome in prenatally diagnosed cystic lung lesions. J Pediatr Surg. 2002;37(3):331–338. ↩
Fetal Ultrasound: A Critical Tool in Diagnosing Occipital Encephalocele
The journey of pregnancy is often filled with anticipation and joy, but it can also present unexpected challenges. One such challenge is the prenatal diagnosis of congenital anomalies, with neural tube defects (NTDs) being among the most significant. Among these, the occipital encephalocele stands out as a distinct entity, and fetal ultrasound (USG) remains the cornerstone for its early and accurate detection.
Understanding Occipital Encephalocele
An encephalocele is a type of neural tube defect where there's a protrusion of brain tissue, meninges (the protective membranes surrounding the brain), or both, through an opening in the skull. When this defect occurs at the back of the head, it's termed an occipital encephalocele – the most prevalent form of this anomaly. These defects arise from a failure of the neural tube to close completely during early fetal development.
The Role of Fetal Ultrasound
Fetal ultrasound offers a non-invasive, real-time window into the developing fetus, making it invaluable for diagnosing congenital conditions. For occipital encephalocele, ultrasound can identify characteristic features, often as early as the first trimester (11-14 weeks) and with high confidence during the routine second-trimester anomaly scan.
Cranial Bony Defect: The hallmark of an encephalocele is a visible gap or defect in the occipital bone (at the back of the skull). This defect can vary significantly in size, from a small opening to a large breach.
Protruding Sac-like Mass: Extending from this skull defect, a sac-like mass will be observed. The nature of this mass is crucial for prognosis:
Meningocele: If the sac contains only cerebrospinal fluid (CSF) and meninges, it appears purely cystic on ultrasound.
Encephalocele/Meningoencephalocele: If the sac contains brain tissue (with or without meninges and CSF), it will have solid or mixed cystic and solid components. The presence of identifiable brain tissue within the sac is a critical finding for assessing severity.
Continuity with Intracranial Contents: The protruding mass will demonstrate continuity with the brain structures inside the skull.
Distortion of Intracranial Anatomy: Depending on the size of the encephalocele and the amount of brain tissue herniated, the normal intracranial landmarks (such as the ventricles, thalami, and cerebellum) may appear displaced or distorted.
Beyond the Primary Diagnosis: Associated Findings
A thorough fetal ultrasound examination for occipital encephalocele goes beyond merely identifying the primary defect. It's imperative to actively search for associated anomalies, as these significantly impact prognosis and management:
Hydrocephalus: The most common associated finding, characterized by an excessive accumulation of CSF within the brain's ventricles, leading to their enlargement.
Microcephaly:An abnormally small head size, which often indicates significant brain involvement and a poorer outcome.
Agenesis of the Corpus Callosum: Absence or underdevelopment of the major nerve tract connecting the two cerebral hemispheres.
Chiari Malformations & Dandy-Walker Malformation: Other brain malformations that can coexist.
Other Neural Tube Defects: Such as spina bifida, though less common with isolated occipital encephaloceles.
Systemic Anomalies: Encephaloceles can be part of broader genetic syndromes.For instance, in Meckel-Gruber Syndrome, occipital encephalocele is typically seen alongside polydactyly (extra fingers or toes) and multicystic dysplastic kidneys.Other syndromes like Walker-Warburg syndrome and Amniotic Band Syndrome can also be associated.
Polyhydramnios: An excess of amniotic fluid, which can sometimes be an indirect sign of a fetal anomaly affecting swallowing.
Differentiating from Other Conditions
While occipital encephalocele has distinct ultrasound features, it's essential to differentiate it from other conditions that might present as an extracranial mass at the back of the head:
Cystic Hygroma: Typically appears as a multi-loculated cystic mass, often bilateral, and importantly, without an underlying skull defect or connection to the brain.
Scalp Edema/Hydrops: Generalized swelling of the fetal scalp due to fluid retention, usually accompanied by fluid accumulation in other body cavities.
Cranial Teratoma: These are tumors that can be solid, cystic, or mixed, but generally do not show continuity with intracranial structures through a skull defect.
Prognostic Implications and Management
The prognosis for a fetus diagnosed with occipital encephalocele is highly variable and depends heavily on:
Contents of the Sac: Prognosis is significantly poorer if the sac contains brain tissue compared to a meningocele (CSF and meninges only). Brain tissue herniation is associated with higher mortality and severe neurological impairments.
Size of the Encephalocele:Larger defects and more extensive herniation generally lead to worse outcomes.
Presence of Microcephaly: This is a strong indicator of severe brain involvement and poor neurodevelopmental prognosis.
Associated Anomalies: The presence of hydrocephalus, other brain malformations, or involvement in a syndromic condition significantly worsens the outlook.
Upon diagnosis, a multidisciplinary team approach is crucial. This typically involves:
Maternal-Fetal Medicine Specialists: For ongoing monitoring and pregnancy management.
Fetal MRI: Often recommended to provide a more detailed anatomical assessment of the brain and the contents of the encephalocele, aiding in surgical planning and prognostication.
Genetic Counselors: To discuss the possibility of underlying chromosomal abnormalities or genetic syndromes, and to offer options for genetic testing (e.g., amniocentesis for karyotyping and microarray).
Pediatric Neurosurgeons: For counseling on potential postnatal surgical repair and anticipated neurological outcomes.
Neonatologists: For planning immediate postnatal care.
Parents receive comprehensive counseling regarding the severity of the condition, potential long-term outcomes, and available management options, which may range from continuing the pregnancy with planned postnatal intervention to considering termination of pregnancy in severe cases, especially when diagnosed early.
Conclusion:
Fetal ultrasound serves as an indispensable tool in the early and accurate diagnosis of occipital encephalocele. Its ability to provide detailed anatomical information is critical for guiding prenatal counseling, assessing prognosis, and planning appropriate perinatal and postnatal management. While the diagnosis can be challenging for expectant parents, timely detection through advanced imaging allows for informed decision-making and optimal care for the affected fetus and family.
Ventricular Septal Defect in Fetal Echocardiography: Assessment Across Multiple Views
Abstract:
Ventricular septal defect (VSD) is one of the most common congenital heart anomalies detected during fetal echocardiography. Accurate prenatal diagnosis is crucial for parental counseling, perinatal management, and planning postnatal care. This article reviews the sonographic appearance of VSDs in different echocardiographic views, emphasizing the importance of a systematic approach and the utility of various planes in improving diagnostic accuracy.
Introduction
Ventricular septal defects are characterized by an opening in the interventricular septum, allowing abnormal communication between the left and right ventricles. VSDs can be isolated or associated with complex congenital heart disease. Prenatal detection is most often performed during a detailed fetal anatomy scan between 18 and 24 weeks of gestation using fetal echocardiography.
Multiple imaging planes are needed to confirm the diagnosis and rule out false positives due to dropouts or artifacts. Understanding the orientation of the interventricular septum in different views is essential.
Classification of VSDs
VSDs are classified based on their location within the septum:
Perimembranous (most common)
Muscular (mid-muscular, apical, or anterior/posterior)
Inlet (associated with atrioventricular canal defects)
Outlet (subpulmonary or supracristal)
Fetal Echocardiographic Views for VSD Evaluation
1. Four-Chamber View
Utility: Primary screening view.
Findings: May reveal mid-muscular or apical VSDs.
Limitations: Membranous and outlet VSDs may be missed due to septal orientation and dropouts.
Tips: Adjust gain settings and use color Doppler to differentiate true defects from artifacts.
2. Left Ventricular Outflow Tract (LVOT) View
Utility: Ideal for identifying perimembranous and outlet VSDs.
Findings: Look for continuity between the ventricular septum and aortic root; disruption suggests a VSD.
Color Doppler: Reveals left-to-right shunting during systole.
3. Right Ventricular Outflow Tract (RVOT) View
Utility: Helps identify outlet VSDs.
Findings: Evaluate the relationship between the VSD and pulmonary valve.
Clinical Tip: Outlet VSDs can be associated with conotruncal anomalies like Tetralogy of Fallot.
4. Three-Vessel View and Three-Vessel Trachea View
Utility: Not typically used to directly visualize VSDs but helpful in identifying associated anomalies (e.g., conotruncal defects).
Findings: Abnormal size or alignment of great vessels may suggest associated outflow tract anomalies.
5. Short-Axis View of the Ventricles
Utility: Complements four-chamber view in detecting muscular VSDs.
Technique: Use a transverse plane through the fetal chest to identify muscular septal discontinuities.
6. Sagittal and Subcostal Views
Utility: Helpful in detecting apical and posterior muscular VSDs.
Technique: Align the transducer to follow the long axis of the heart for optimal visualization of the septum.
Role of Color and Power Doppler
Color Doppler is essential in confirming VSDs by identifying abnormal flow between the ventricles. High frame rates and small color boxes improve resolution. Power Doppler can be helpful in low-velocity shunts or small defects not seen on standard color Doppler.
Pitfalls and Artifacts
Dropout artifact: A common mimic of VSD, particularly in the membranous septum.
Angle dependency: Some defects are only visible in specific planes.
Small defects: May be missed without meticulous scanning and appropriate Doppler settings.
Clinical Implications
Prenatal identification of a VSD allows for:
Evaluation for additional cardiac anomalies
Genetic counseling (as VSDs may be associated with chromosomal abnormalities, especially in large or multiple defects)
Perinatal planning (e.g., delivery at a center with neonatal cardiac care)
Postnatal follow-up: Some small muscular VSDs close spontaneously; others may require surgical intervention.
Conclusion
Ventricular septal defects can be detected with high sensitivity using a multi-view approach in fetal echocardiography. Each echocardiographic plane provides unique insight into different parts of the septum. Combined with color Doppler imaging, this comprehensive approach improves diagnostic accuracy and supports optimal clinical decision-making.
Congenital diaphragmatic hernias are seen in 1 of every 2000-4000 live births. 84% are left-sided, 13% are right-sided and 2% bilateral 6.
Clinical presentation
Most congenital diaphragmatic hernias are detected either soon after birth or on antenatal ultrasound. Mortality is predominantly due to the development of pulmonary hypoplasia, which is thought to be due to the mass effect on the developing lung. Such neonates are hypoxic and have persistent fetal circulation due to pulmonary hypoplasia and pulmonary hypertension.
Pathology
Diaphragmatic development is usually complete by ~9th week of gestation. Congenital diaphragmatic hernias result from failure of fusion of one of the pleuroperitoneal canals at about 8 weeks gestation. They may contain the stomach, intestines, liver, or spleen.
Types
Congenital diaphragmatic herniation can be grouped into two basic types on location:
Bochdalek hernia
most common fetal congenital diaphragmatic hernia
more common on the left: 75-90%
posterolateral
large and associated with poorer outcome
presents earlier
mnemonic: BBBBB
Morgagni hernia
less common
anterior
presents later
Associations
While a CDH can occur as an isolated condition, associated anomalies are relatively common and include:
pulmonary hypoplasia: also a complication
left ventricular hypoplasia due to impaired umbilical venous return 19
early ventricular dysfunction, about 40% 18
intestinal malrotation: in up to 45% of cases 17
bronchopulmonary sequestration
aneuploidy: can be present in up to 50% of cases ref
trisomy 13
trisomy 18
trisomy 21
Turner syndrome: monosomy X
Pallister-Killian syndrome: tetrasomy 12p
Fryns syndrome
Cornelia de Lange syndrome
congenital cardiac anomalies
neural tube defects
anencephaly
spina bifida
Radiographic features
Plain radiograph
indistinct diaphragm with opacification of part of or all the hemithorax (typically left sided)
scaphoid abdomen
deviation of lines 3
endotracheal tube
nasogastric tube
umbilical arterial and venous catheters
Antenatal ultrasound
Indirect sonographic findings that should prompt a search for CDH include 7:
polyhydramnios
cardiomediastinal shift +/- abnormal cardiac axis
inability to demonstrate the normal stomach bubble
The study should be performed in the true transverse plane. Sonographic diagnosis of CDH can be made from the following findings 7,8:
absent bowel loops in the abdomen
intrathoracic herniation of the liver; noted in up to 85% of cases and is associated with a worse prognosis
peristaltic bowel movements in the chest
herniation into the chest may occur intermittently
abdominal circumference is reduced (due to herniation of organs)
left-sided CDH
stomach and small bowel (echo-free) at the same transverse level as the heart on four-chamber view: this makes left sided hernias comparatively easier to detect on ultrasound (as opposed to herniation of echogenic liver on the right side)
stomach and small bowel superior to the inferior margin of the scapula
leftward displacement of the gallbladder
right-sided CDH
color Doppler study
leftward bowing of the umbilical segment of the portal vein
portal branches to the lateral segment of the left hepatic lobe coursing towards or above the diaphragm
gallbladder present above the diaphragm
echogenic space between the left heart border and stomach representing the left hepatic lobe
Although classically considered a cystic echogenic lung mass, there are reports of CDH appearing initially as a solid echogenic lung mass that evolves in appearance with advancing gestation 9.
The observed-to-expected lung-to-head ratio (O/E LHR) may be calculated and correlates with the degree of pulmonary hypoplasia. Studies suggest that the degree of lung hypoplasia can be used to predict survival rates and the numbers from the Antenatal-CDH-Registry group that apply to isolated left-sided CDH and liver herniation are shown below 10,11:
O/E LHR <15% (extreme pulmonary hypoplasia): virtually no chance of survival
lungs appear hyperintense (composed primarily of water) while heart, mediastinum and liver appear hypointense
Lung-head ratio: can be assessed on both ultrasound or MRI; MRI measured LHR has been found to have a slightly higher prognostic accuracy than with ultrasound 5.
MRI allows the measurement of fetal lung volumes which provide an estimate of the severity of pulmonary hypoplasia. The total fetal lung volume (TFLV) can be calculated from a contiguous T2-weighted HASTE sequence and an observed-to-expected TFLV (O/E TFLV) derived. It has been found to predict well both mortality and morbidity, including the need for ECMO and the development of bronchopulmonary dysplasia 14,15.
Treatment and prognosis
Fetuses with an antenatal diagnosis of CDH should be delivered in a tertiary referral center with access to neonatal intensive care and pediatric surgical facilities.
Large CDH have a poor prognosis, due to pulmonary hypoplasia and perinatal mortality may be as high as 80%. Successful management is dependent on specialist pediatric facilities, with the ability to offer surgery, ECMO etc.
early ventricular dysfunction especially biventricular dysfunction 18
the presence of associated abnormalities
bilateral CDH
unfavorable lung: head ratio
A composite prognostic index (CDH-CPI) comprising 10 prenatal parameters has been developed and was found to have a stronger correlation with survival and need for ECMO than any one parameter individually 16.
Some centers perform in utero surgery in selected cases. One such surgical approach is the fetoscopic endotracheal occlusion (FETO) procedure, in which the fetal trachea is temporarily occluded by a balloon to allow expansion of the fetal lungs with fluid and consequently prevent some degree of pulmonary hypoplasia 21. A randomized controlled trial demonstrated a survival benefit in severe CDH 21.
Complications
development of pulmonary hypoplasia
development of pulmonary hypertension
severity may be predicted by the modified McGoon index
To describe a new sign of cleft lip and palate (CLP), the maxillary gap, which is visible in the mid-sagittal plane of the fetal face used routinely for measurement of nuchal translucency thickness.
Methods
This was a retrospective study of stored images of the mid-sagittal view of the fetal face at 11–13 weeks' gestation in 86 cases of CLP and 86 normal controls. The images were examined to determine if a maxillary gap was present, in which case its size was measured.
Results
In 37 (43.0%) cases of CLP the defect was isolated and in 49 (57.0%) there were additional fetal defects. In the isolated CLP group, the diagnosis of facial cleft was made in the first trimester in nine (24.3%) cases and in the second trimester in 28 (75.7%). In the group with additional defects, the diagnosis of facial cleft was made in the first trimester in 46 (93.9%) cases and in the second trimester in three (6.1%). A maxillary gap was observed in 96% of cases of CLP with additional defects, in 65% of those with isolated CLP and in 7% of normal fetuses. There was a large gap (>1.5 mm) or complete absence of signals from the maxilla in the midline in 69% of cases of CLP with additional defects, in 35% of those with isolated CLP and in none of the normal controls.
Cleft lip and palate (CLP) is a common congenital defect that can be either isolated or associated with a wide range of chromosomal abnormalities and genetic syndromes. Isolated cleft lip or cleft palate may escape prenatal detection, but combined CLP is diagnosed easily during the routine second-trimester scan1, 2.
In the last decade, widespread use of the 11–13-week scan in screening for aneuploidies with measurement of fetal nuchal translucency (NT) thickness has resulted in many major fetal defects being diagnosed in the first trimester of pregnancy3. This can be achieved by direct visualization of the fetal anatomy, for example in cases of anencephaly, exomphalos and megacystis3, or by focused assessment of specific fetal structures triggered by the finding of indirect signs, for example increased NT in association with major cardiac defects4, 5 and abnormal intracranial translucency (IT) in association with open spina bifida6-9.
Most cases of CLP are not detected during the first-trimester scan3. Improved detection may be achieved by targeted examination of the face in a coronal view and assessment of the retronasal triangle10. However, the uptake of such assessment may be limited by the necessity to include examination of a plane additional to the standard mid-sagittal view that is necessary for measurement of fetal crown–rump length, NT, nasal bone (NB) and IT.
The objectives of this study were first, to describe a new sign of CLP, the maxillary gap, visible in the mid-sagittal plane of the fetal face, which is used routinely for measurement of NT, NB and IT (Figures 1 and 2) and, second, to provide preliminary data on the potential value of the maxillary gap in the early diagnosis of CLP.
Standard mid-sagittal view of the fetal face used routinely at 11–13 weeks, illustrating the maxilla in a normal fetus (a) and the maxillary gap (open arrow) in a fetus with cleft lip and palate (b).
Ultrasound images in the mid-sagittal plane of the face in a normal fetus with no maxillary gap (a), a normal fetus with small maxillary gap (arrow) (b) and four fetuses with cleft lip and palate, showing partial (c,d,e) or complete (f) maxillary gap (arrows).
SUBJECTS AND METHODS
In three cases of CLP we made an incidental observation that, in the mid-sagittal view of the fetal face at 11–13 weeks' gestation, there was a gap in the echogenic line representing the palate; no such gap was observed in 10 consecutive cases of normal fetuses. We therefore subsequently conducted a search of the databases of two major centers of prenatal diagnosis, to identify fetuses with CLP which underwent routine first-trimester screening for aneuploidies. The stored images of the mid-sagittal view of the face in these cases were retrieved and for each case a control was selected: a fetus examined on the same day that was subsequently liveborn without any abnormality.
The diagnosis of CLP was made in a coronal view of the face. In most cases which underwent termination of pregnancy, diagnosis was confirmed clinically or at autopsy. Cases with isolated cleft lip or cleft of the posterior palate were not included in the study. Transabdominal ultrasound examinations were performed initially as part of routine NT screening and, in the presence of fetal malformations, an additional transvaginal examination was performed selectively.
All retrieved images of cases and controls were assessed offline by two independent examiners (R.C., G.O.) who were unaware of the final diagnosis. The images were examined to determine if a maxillary gap was present; if this was the case we determined whether it was partial or complete (Figure 2) and measured the size of the gap. Ultrasound findings and pregnancy outcome data were obtained from the databases and their relation to the maxillary gap was examined.
RESULTS
The study population comprised 86 cases of CLP with satisfactory images of the mid-sagittal view of the fetal face at 11–13 weeks, identified from the combined databases of the two centers. The characteristics of the study population and controls are summarized in Table 1. In the controls no defect was detected in either the first- or the second-trimester scan and all pregnancies resulted in healthy live births with no defects.
Table 1. Characteristics of study group of 86 fetuses with facial cleft and 86 normal controls
Characteristic
Normal controls (n = 86)
Facial cleft
Isolated (n = 37)
Other defects (n = 49)
Maternal age (years)
32 (30–36)
32 (26–35)
34 (28–39)
Gestational age (weeks)
12.6 (12.3–12.7)
13.0 (12.7–13.3)
12.7 (12.1–13.3)
Crown–rump length (mm)
61.0 (57.3–64.0)
68.3 (63.5–72.9)
60.2 (54.6–68.3)
Nuchal translucency thickness (mm)
1.6 (1.4–1.9)
1.9 (1.7–2.1)
2.8 (1.9–4.9)
Fetal karyotype
Normal or normal live birth
86 (100)
37 (100)
12 (24.5)
Trisomy 13
—
—
23 (46.9)
Trisomy 18
—
—
9 (18.4)
Other
—
—
3 (6.1)
Unknown
—
—
2 (4.1)
Cleft location
Median
—
—
12 (24.5)
Bilateral
—
7 (18.9)
26 (53.1)
Unilateral
—
30 (81.1)
11 (22.4)
Maxillary protrusion
—
7 (18.9)
20 (40.8)
Time of diagnosis
First trimester
—
9 (24.3)
46 (93.9)
Second trimester
—
28 (75.7)
3 (6.1)
Outcome
Live birth
86 (100)
33 (89.2)
2 (4.1)
Neonatal death
—
1 (2.7)
—
Termination of pregnancy
—
3 (8.1)
45 (91.8)
Fetal death
—
—
2 (4.1)
Data are given as median (interquartile range) or n (%).
The diagnosis of facial cleft was made in the first trimester in 55 (64.0%) and in the second trimester in 31 (36.0%) of the CLP cases (Table 1). The diagnosis was suspected in the first trimester in some cases due to maxillary protrusion or to the presence of associated anomalies and was confirmed by examining a coronal view of the face and the oblique view of the retronasal triangle.
In 37 (43.0%) cases the facial cleft was an isolated finding; in three (8.1%) of these the pregnancy was terminated at the request of the parents, one (2.7%) underwent neonatal death due to birth asphyxia and in 33 (89.2%) the pregnancy resulted in live birth and the infant underwent surgery for correction of the defect. In this group with isolated CLP the diagnosis of facial cleft was made in the first trimester in nine (24.3%) cases and in the second trimester in 28 (75.7%).
In 49 cases of CLP there were additional fetal defects. In 47 cases fetal karyotyping following chorionic villus sampling or amniocentesis was carried out; the karyotype was normal in 12 (25.5%) and abnormal in 35 (74.5%), including 23 cases of trisomy 13, nine of trisomy 18 and three other chromosomal abnormalities. In 45 cases the pregnancy was terminated at the request of the parents, two fetuses died in utero and in two the pregnancy resulted in live birth and the infant underwent surgery for correction of the defect. In this group with additional defects the diagnosis of facial cleft was made in the first trimester in 46 (93.9%) cases and in the second trimester in three (6.1%).
A maxillary gap was observed in six (7.0%) of the controls, in 24 (64.9%) cases with isolated CLP and in 47 (95.9%) cases of CLP with additional abnormalities (Table 2). Figure 2 illustrates partial and complete maxillary gaps. The size of the gap in all six controls was < 1.5 mm. Of the 24 cases of isolated CLP with a maxillary gap, its size was < 1.5 mm in 11 (45.8%) and 1.5–5.0 mm in 13 (54.2%). Of the 47 cases with CLP, additional defects and a maxillary gap, the gap size was < 1.5 mm in 13 (27.7%), 1.5–5.0 mm in 21 (44.7%) and complete in 13 (27.7%); in eight of the latter fetuses there was holoprosencephaly and a median facial cleft.
Table 2. Type and size of maxillary gap in 86 fetuses with facial cleft and 86 normal controls
Maxillary gap characteristic
Normal controls (n = 86)
Facial cleft
Isolated (n = 37)
Other defects (n = 49)
Type
No gap
80 (93.0)
13 (35.1)
2 (4.1)
Partial gap
6 (7.0)
24 (64.9)
34 (69.4)
Complete gap
—
—
13 (26.5)
Size
< 1.5 mm
6 (100)
11/24 (45.8)
13/47 (27.7)
1.5–5 mm
—
13/24 (54.2)
21/47 (44.7)
Complete
—
—
13/47 (27.7)
Data are given as n (%).
Protrusion of the maxilla in the profile view was observed in 20 (40.8%) of the 49 fetuses with CLP and additional abnormalities and in seven (18.9%) of the 37 cases with isolated CLP (Table 1).
DISCUSSION
This study demonstrates that CLP can be suspected at 11–13 weeks' gestation in the presence of a maxillary gap in the standard mid-sagittal view of the fetal face that is used routinely in screening for chromosomal abnormalities. A maxillary gap was observed in 96% of cases of CLP with additional abnormalities, in 65% of those with isolated CLP and in 7% of normal fetuses. There was a large gap or complete absence of signals from the maxilla in the midline in 69% of cases of CLP with additional abnormalities, in 35% of those with isolated CLP and in none of the normal controls.
At the end of complex embryological development of the midline of the face between 6 and 12 weeks' gestation, the nose and lip are formed and the palate closes. Anatomically, the palate is then composed of three parts: the anterior or primary palate, including the alveolar ridge, the posterior or secondary palate and the soft palate11. At the time of the 11–13-week scan, some parts of the maxilla may still show lack of ossification. This may explain the finding of a gap in some of the normal controls: in all of these, the size of the suspected gap was very small (< 1.5 mm). Therefore, suspicion of a small maxillary gap in the mid-sagittal view in the presence of an intact maxilla in the coronal view can be considered as a normal finding.
The diagnosis of CLP was made at the 11–13-week scan in 94% of the cases with other abnormalities, but in only 24% of those with isolated CLP. This is not surprising because, in the presence of often multiple sonographic defects, a systematic search is undertaken for additional defects, including CLP, especially in cases with suspected trisomy 18 or 13.
Examination of the maxilla in the mid-sagittal view of the fetal face at 11–13 weeks has been reported previously, in studies which focused mainly on its length in normal fetuses and in fetuses with trisomy 21 or on its use as a landmark for calculation of the frontomaxillary facial angle12-15. Our present study emphasizes a new aspect of early assessment of the maxilla: looking for a maxillary gap as a potential marker of CLP.
In the prenatal diagnosis of facial cleft the typical recommended views during the second-trimester scan are the axial view of the maxilla and the coronal view of the nose and lip11, 16, 17. The importance of the mid-sagittal view of the palate was reported recently for demonstration of the so-called ‘equals sign’ corresponding to the anatomical appearance of the soft palate and uvula18. It was suggested that, in the first trimester, identification of cleft palate necessitates a coronal view of the anterior bony face and demonstration of the retronasal triangle10. Some studies have reported on the use of the retronasal triangle view with three-dimensional (3D) ultrasound to demonstrate CLP and retrognathia19-22. However, the retronasal triangle plane on two-dimensional (2D) or on 2D in combination with 3D ultrasound has not been accepted widely as a standard view in routine screening. In contrast, we have highlighted the importance of the maxillary gap as a marker of possible CLP because the marker is visible in the standard plane that is obtained routinely in screening for aneuploidies.
The main strength of the study is the large number of affected cases with stored images of quality acceptable for assessment. The main limitation relates to its retrospective nature and reliance on stored images. Although for the cases of CLP detected in the first trimester there were numerous images and video recordings, for those in which the diagnosis was missed there were only a few stored images for assessment.
In conclusion, examination of the mid-sagittal view of the fetal face, which is performed routinely for assessment of fetal NT, NB and the posterior brain region, can identify a maxillary gap and lead to first-trimester diagnosis of CLP. In cases of maxillary gap, the sonographer should undertake detailed examination of the face and palate regions. Prospective large studies are necessary to determine the performance of the maxillary gap in screening for facial clefts.